

Technology Transfer in Pharma: Best Practices for Successful CMC and Regulatory Compliance
The Transformative Power of AI-Driven CMC Writing in Modern Pharmaceutical Development

25 Jul, 2025
Meeting the Demands of Modern CMC Documentation
Chemistry, Manufacturing, and Controls (CMC) documentation is one of the most demanding elements of any regulatory submission. Technical writers have to translate detailed manufacturing data – batch formulations, process controls, stability studies into a coherent dossier that regulators can evaluate. In today’s environment, where biologics, biosimilars, and cell- and gene-based therapies are on the rise, the sheer volume and complexity of data can overwhelm traditional writing processes.
Teams often juggle dozens of spreadsheets, hundreds of analytical reports, and multiple site-specific protocols. Every addition or revision must be incorporated into the final dossier with perfect consistency. Mistakes can lead to questions, delays, or even outright rejection. Consequently, regulatory departments are under intense pressure to deliver error-free CMC modules on aggressive timelines.
Journal of Pharmaceutical Sciences reports that 85% of FDA delays originate from CMC deficiencies, while Tufts Center for the Study of Drug Development quantifies each day’s delay at $2M in lost revenue. This documentation challenge intensifies with novel therapies like cell/gene treatments where manufacturing variability directly impacts clinical outcomes.
With such high stakes, pharmaceutical companies can no longer rely on manual processes alone – leading to a surge in interest in AI-powered solutions.
Why Traditional CMC Methods No Longer Work
The rise of biologics, ATMPs, and biosimilars has made old approaches to CMC documentation obsolete. These therapies create new challenges that manual processes can’t handle:
-
Manufacturing Defines the Product
For biologics, quality depends entirely on production controls. Documentation must include:
- Full process details from cell lines to final product
- Viral clearance proof
- Molecular structure analysis
Standard small-molecule templates miss these requirements.
-
More Data, More Rules
Regulators now want:
- Different details for each region (FDA vs EMA vs PMDA)
- Hundreds of quality measurements per batch
- Patient-specific records for cell therapies
-
Manual Systems Can’t Keep Up
Where simple Word files once worked, now:
- Plants in different countries must show identical results
- Every patient’s treatment needs unique paperwork
- Small process changes require full document updates
The Solution:
Old methods using separate files and manual reviews can’t meet these demands. AI-powered systems are now essential for compliant, efficient documentation.
AI and Automation: The New Frontier in Regulatory Writing
Forward-thinking organizations are leveraging technology to address these challenges through:
Cross-Dossier Management Tools
Cloud-based eCTD solutions now enable version control across global submissions while automatically flagging gaps in comparability studies.
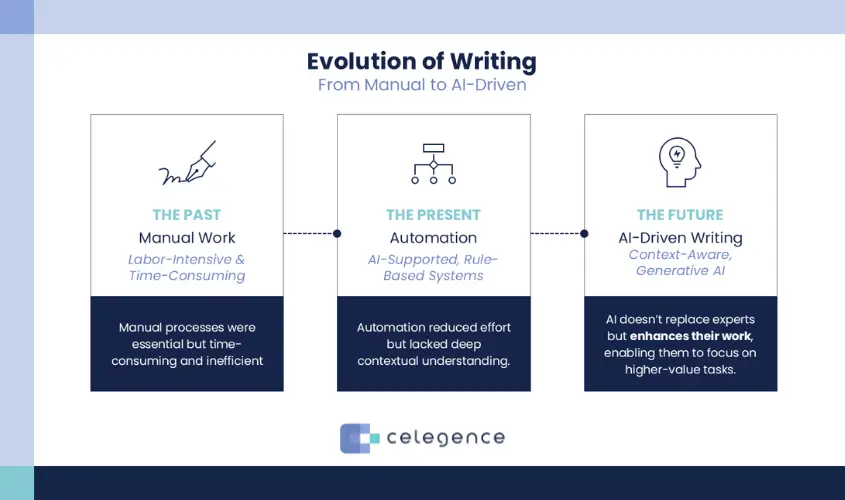
Therapy-Specific Documentation Approaches
- Biologics: Capturing Process Complexity – Monoclonal antibodies and fusion proteins require specialized documentation frameworks that address:
- Viral clearance validation strategies
- Higher-order structure characterization
- Container-closure interaction studies
Leading companies now use template libraries that pre-validate these sections against current FDA QbR expectations.
- ATMPs: Novel Science, Novel Documentation – Cell and gene therapies demand unprecedented CMC rigor, particularly for:
- Closed loop tracking of critical quality attributes
- Real-time ICH Q5A(R2) compliance monitoring
- Patient-specific batch documentation
The EMA’s recent revision of ATMP guidelines underscores how rapidly these requirements evolve.
A Smarter Way to Author CMC Content
Structured authoring platforms address these challenges by combining three pillars:
- Automation of Repetitive Tasks
Rather than manually copying batch details or stability tables, an automated engine extracts and formats data directly from source files. This eliminates transcription errors and frees writers to focus on interpretation and justification. - Purpose-Suitable Templates
Content frameworks built around therapeutic classes – such as monoclonal antibodies or gene therapies – ensure that required sections (viral clearance strategy, comparability summaries, container-closure data) are always included, in the correct order, and to the right level of detail. - Collaborative Review Workflows
Multiple stakeholders – scientists, quality leads, regulatory experts – can comment and approve content simultaneously. Real-time version control prevents overwritten conflicts, and built-in dashboards highlight outstanding comments or pending approvals. By weaving these capabilities into the writing process, teams can shift their focus from document assembly to scientific rigor and regulatory strategy. Errors and rework drop dramatically, and submission packages become more cohesive and defensible.
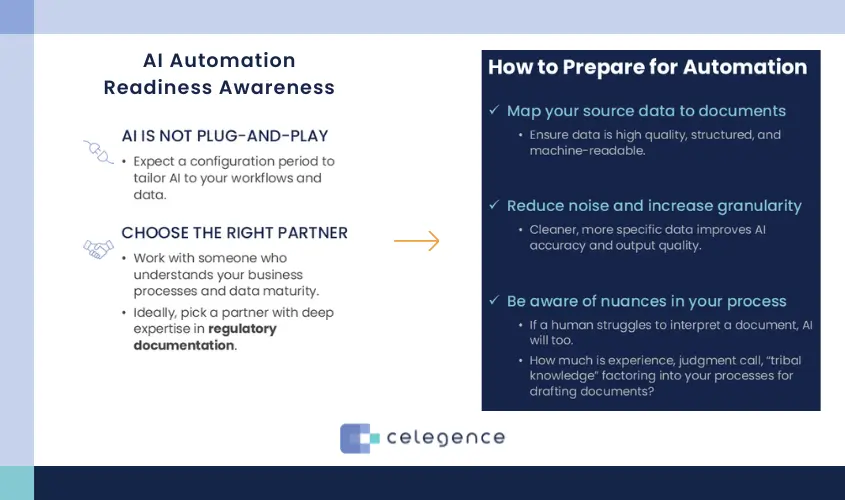
Supporting Documentation for Biologics and ATMPs
The platform’s structured approach shines in complex therapy areas.
For biologics:
- Extensive viral clearance validation sections are pulled into standardized subsections, ensuring nothing is overlooked.
- Higher-order structure assessments – often requiring detailed figures and data tables – are integrated via content modules that manage figure numbering and captions automatically.
- Container-closure compatibility data are formatted according to regional requirements, whether for FDA, EMA, or PMDA.
For ATMPs, the challenge is even greater:
- Patient-specific batch summaries require unique data handling for each lot.
- Critical Quality Attributes (CQAs) must be linked to manufacturing controls, analytical methods, and stability results – often across multiple documents.
- Emerging guidance like ICH Q5A(R2) necessitates rapid template updates as new regulatory expectations appear.
By codifying these therapy-specific requirements into configurable templates, structured authoring ensures that both the content and its presentation remain fully compliant and readily auditable.
Real World Case Study
A global Pharmaceutical company faced regular document crashes, formatting inconsistencies, and lengthy review cycles when drafting regulatory submissions. After transitioning to a structured authoring environment powered by an AI engine, the team reported the following benefits:
- Seamless, crash-free document editing across dozens of contributors
- Standardized reference and glossary management, removing citation errors
- Parallel review capabilities, reducing internal review timelines by nearly half
AI-driven platforms have been applied to both pharmaceutical CMC and MedTech examples with comparable efficiency gains – especially in managing multiple sites, dozens of batch reports, and evolving global standards.
Getting Started: A Phased Approach
With regulatory scrutiny intensifying, delaying AI adoption risks costly submission delays. Here’s how to begin:
- Select High-Value Pilot Sections: Choose content types that consume the most manual effort – impurity profiles, stability summaries, or comparability protocols – to demonstrate rapid return on investment.
- Import and Map Legacy Documents: Bring existing Word files into the new environment, mapping their structure to built-in templates. This preserves historical data while enforcing new standards.
- Onboard Cross-Functional Teams Together: Train regulatory writers, subject matter experts, and quality reviewers in parallel to align expectations and minimize transition friction.
- Leverage Dashboards for Continuous Improvement: Use real-time metrics to monitor authoring speed, review queue lengths, and consistency issues. Iterate templates and workflows based on these insights.
This phased rollout delivers immediate wins and builds organizational confidence, paving the way for broader adoption across all CMC content areas.
Transforming CMC Documentation into a Competitive Edge
CMC documentation is more than a compliance checkbox – it can drive real business value. By adopting structured authoring platforms that blend automation, therapy-specific templates, and collaborative review, teams gain:
- Faster Time to Submission
Automated data capture and parallel reviews cut down rework and help meet tight deadlines. - Improved Submission Quality
Consistent formatting, accurate calculations, and full audit trails reduce regulator queries. - Scalable Operations
As pipelines grow, structured workflows handle larger data volumes without a proportional increase in headcount. - Enhanced Collaboration
Real-time commenting and version control brings manufacturing, quality, and clinical experts together seamlessly.
Embedding these capabilities turns CMC writing from a bottleneck into a strategic asset—supporting faster market access and stronger global compliance.

Ready to turn CMC writing from a bottleneck into a strategic advantage? Explore Celegence’s AI-driven solutions today, visit celegence.com or contact info@celegence.com.
Other Related Articles

04 Aug, 2026

15 Jul, 2026

02 Jul, 2026

26 Jun, 2026

05 Jun, 2026

