
Beyond “Inactive”: Mastering Type IV DMFs for Excipients Using IPEC Guidance
EU Regulatory Challenges and Considerations in Radiopharmaceuticals

24 Sep, 2025
This is the 3rd blog in our series “Understanding Radiotherapeutics and Radiodiagnostics.” Read our Part 1: Understanding the Regulatory Considerations for Radiotherapeutics and Radiodiagnostics and Part 2 here: US Regulatory Considerations for Radiotherapeutics & Radiodiagnostics.
The European regulatory environment for RPs is inherently more challenging due to the need to secure regulatory approval from multiple health authorities in different countries.
Clinical Trials in the EU
Generally, sponsors can use the Clinical Trial Information System (CTIS) to submit a Clinical Trial Application (CTA) to run a trial in up to 30 European Economic Area (EEA, Figure 1) countries via a single online application (1).
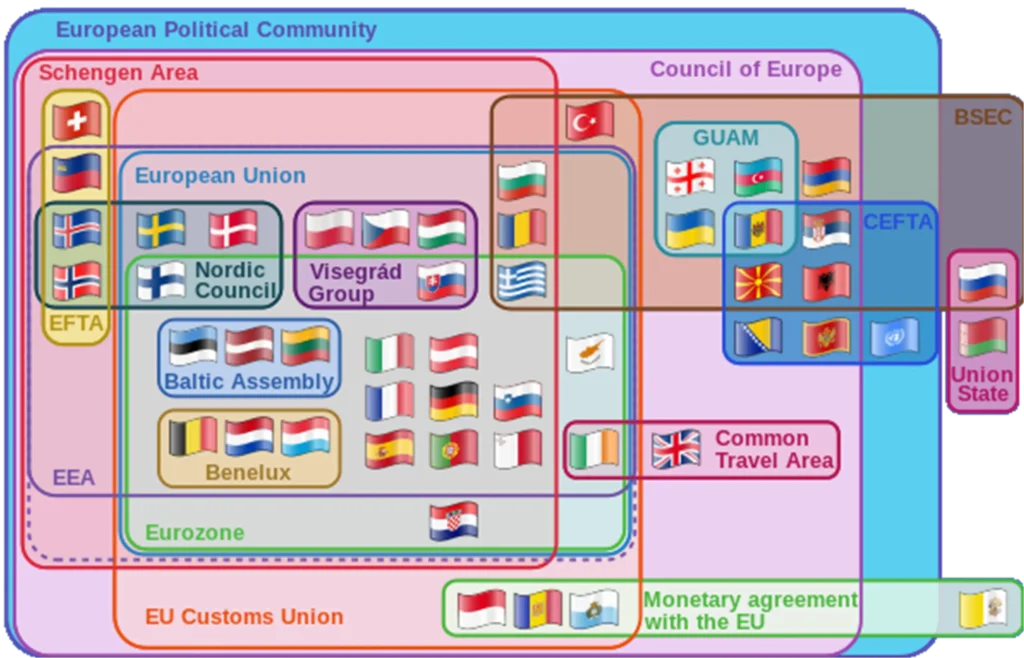
Figure 1: Europe in treaties
Note EEA countries in the purple perimeter
Most countries (except for Bulgaria, Norway, Serbia, and Spain) require additional authorization from their respective national authorities. Health Authorities in some countries (e.g., Belgium, Denmark, Hungary) review separate RP submissions in parallel with CTIS, and thus do not impact the overall review and approval timeline. However, for review processes not conducted in parallel, timelines can vary widely from country to country, from as little as 90 days in the Czech Republic, to as much as seven months in Germany.
Country-Specific Complexities
The following countries impose particularly complex requirements for RP trial applications:
- France: Sites must obtain separate certifications from the Regional Health Agency and the Nuclear Safety Authority.
- Italy: The Italian Ministry of Health (MOH) must be notified 30 days before trial site initiation and after CTIS approval. The additional notification may prolong the overall timeline by a month or more.
- Netherlands: Sites must submit radiation exposure calculations to an internal Radiation Protection Committee. Additionally, a transport license may be required.
- Poland: The CTIS submission dossier must also be sent to the Atomic Energy Agency.
- Sweden: Sites must obtain a license from the Swedish Radiation Protection Authority (SSM).
The intricacies of the European regulatory environment have profound implications for RP clinical trials: if one country has particularly stringent criteria for clinical trial approval, it may deter sponsors from conducting a trial in that country. A sponsor may therefore select another country with less onerous criteria, unless one of the more stringent countries has a specific population the sponsor has targeted for enrolment.
In terms of Radiation Committee (RC) review, the heterogeneity in regulatory processes across countries may impact start-up timelines. Somewhat surprisingly, Spain does not require RC review. Notably, the United Kingdom (UK) regulatory framework is still largely the same as in the EU post-Brexit, in that no special considerations apply to one market versus the other.
Medicinal Product Regulations
Radiopharmaceuticals are classified as medicinal products and are regulated under:
- Directive 2001/83/EC – Governs the manufacture, marketing, and use of medicinal products in the EU (3).
- EU Good Manufacturing Practice (GMP) – Radiopharmaceuticals must comply with EU GMP Guidelines, including Annex 3, which provides specific GMP requirements for radiopharmaceuticals (4).
- Pharmacopoeia Standards – The European Pharmacopoeia (Ph. Eur.) sets quality standards for radiopharmaceuticals (5).
Radiation Protection Laws
Radiopharmaceuticals contain radioactive isotopes, which fall under radiation protection laws:
- Council Directive 2013/59/Euratom (BSS Directive) – Sets safety standards for radiation protection, covering the handling, transport, and disposal of radioactive materials (6).
- Regulations on Transport – EU aligns with IAEA regulations and ADR (for road transport).
Non-Clinical Requirements in the EU
As for non-clinical requirements for RP products in the EU, the EMA outlines a number of scenarios calling for a targeted (in the sense of a reduced) non-clinical program for the non-radioactive part of the RP. Such scenarios may require only partial characterization of the product’s pharmacology, pharmacokinetics (PK), toxicology, genotoxicity, reproductive toxicity, and carcinogenicity for the following types of products (2):
- A known or minimally changed non-radioactive part of a new RP
- A new RP using a microdose approach (Figure 2)
- A new RP using single sub-pharmacological or pharmacologically active doses (Figure 3)
- A new RP using multiple dosing (Figure 4)
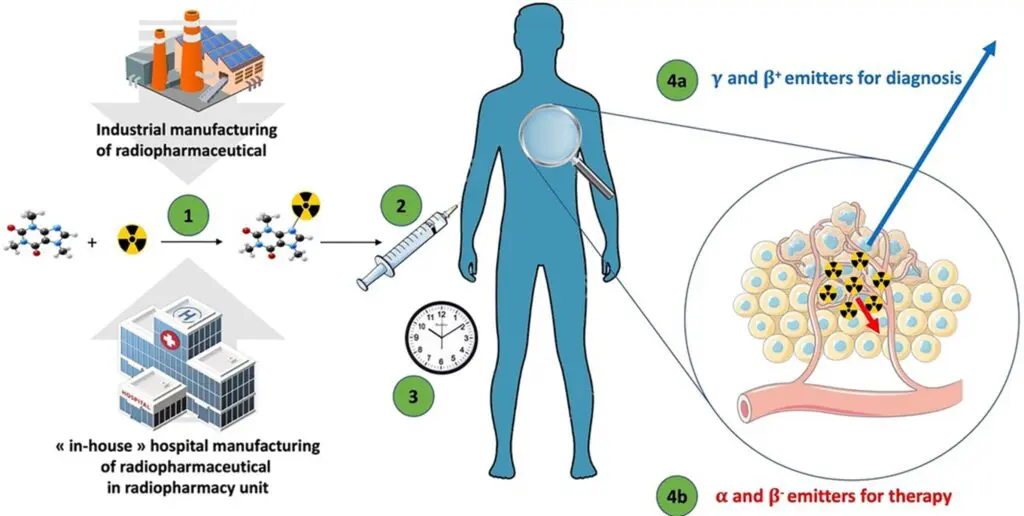
Figure 2: Microdose Radiopharmaceuticals – Microdose radiopharmaceuticals (7).
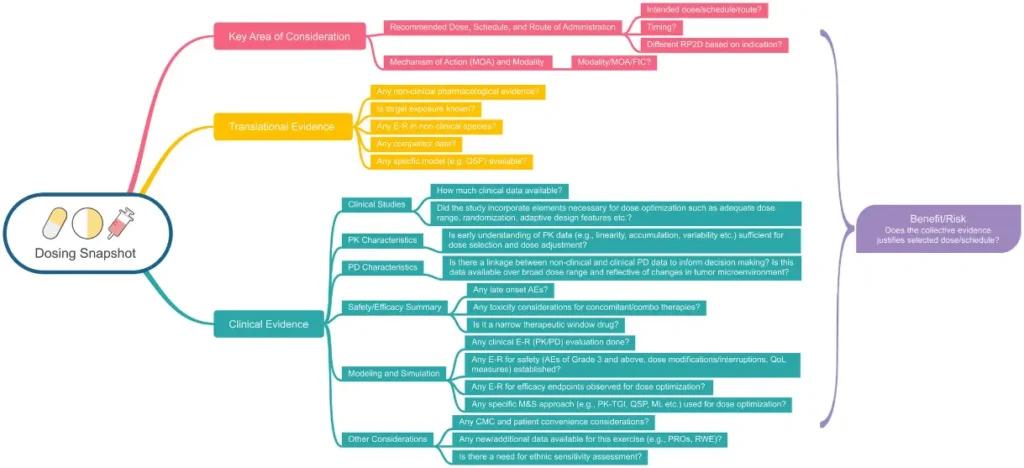
Figure 3: Selection of pharmacologically active doses – Graphical representation of the clinical pharmacology dosing snapshot (9).
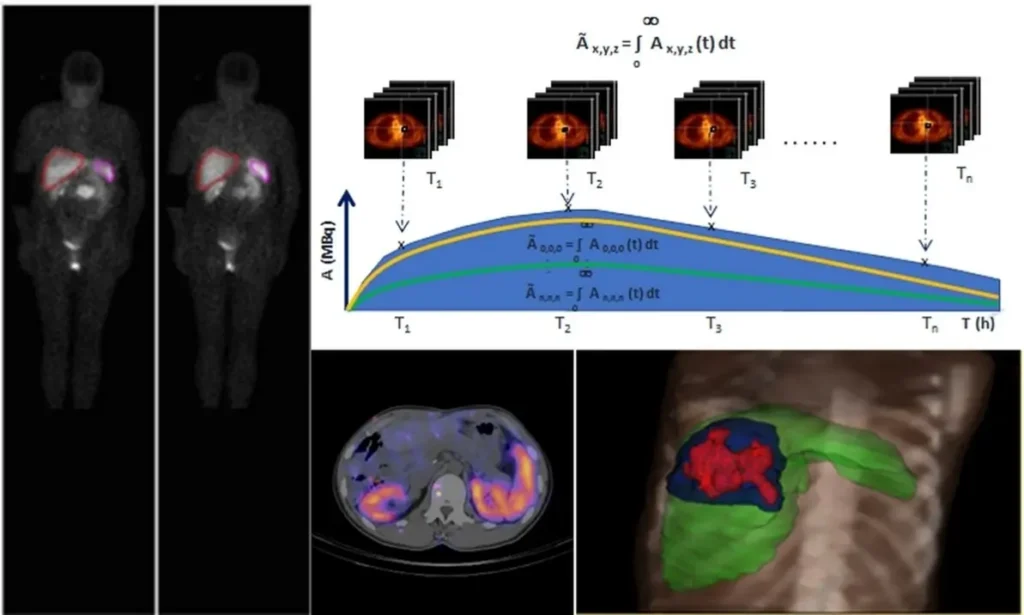
Figure 4: RP using multiple dosing – Uses of Dosimetry in Radiopharmaceutical Therapy (Photo: Dr Massimiliano Pacilio) (10)
The EMA guidance also outlines additional scenarios in which non-clinical testing “could be anticipated” due to “peculiarities.” These include dosages outside microdose, for which “toxicities related to the non-radioactive part are usually still minor compared to radiation-induced toxicities of a radiotherapeutic in oncology treatment.” The amount of non-clinical data required for the non-radioactive part therefore depends on the benefit-risk profile and levels of exposure to the non-radioactive part. Consequently, “a study in one species could be considered to be sufficient” in such cases (1).
Marketing Authorization Pathways
As medicinal products there are two key authorization pathways for radiopharmaceuticals:
- The Centralized Procedure (CP, @EMA) – Required for innovative radiopharmaceuticals used in diagnosis or therapy.
- The Decentralized, Mutual Recognition and National Procedures (via National Competent Authorities, NCAs) – For radiopharmaceuticals provided they do not fall under the mandatory CP, or are produced and used in a single country.
Hospital and Compounding Exemptions
Some radiopharmaceuticals prepared in hospitals or nuclear medicine units (Figure 5) for immediate patient use may be exempt from full marketing authorization but must follow GMP and Euratom safety regulations.
![]()
Figure 5: Nuclear Medicine
In nuclear medicine, very small amounts of radioactive material or radiopharmaceuticals is used for examining organ function or structure for example to diagnose cancer. The radioactive material is absorbed by the soft tissue in the body and helps visualization of abnormalities (11).
Pharmacovigilance and Post-Marketing Surveillance
Regulation (EU) No 1235/2010 (pharmacovigilance of medicinal products for human use, (8)) and Directive 2001/83/EC (as amended by 2010/84/EU) require continuous monitoring of radiopharmaceutical safety. The European Medicines Agency (EMA) oversees pharmacovigilance.
How Celegence can help
Celegence’s team of subject matter experts in regulatory strategy for pharmaceuticals and medical devices ensures that both country-specific and market-wide regulatory requirements are met. We provide sponsors with tailored guidance and develop strategic roadmaps to maximize the efficiency of radiodiagnostics and radiotherapeutics in clinical trials. Most importantly, our experts act as on-the-ground partners, helping sponsors navigate the complexities of the regulatory landscape with confidence.
Contact us today at info@celegence.com to learn how our team can support your radiopharmaceutical programs with confidence and compliance.
References
- Human regulatory: Clinical Trials Information System. European Medicines Agency; 2023. Link.
- Guideline on the non-clinical requirements for radiopharmaceuticals (draft). European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP); 2018. EMA/CHMP/SWP/686140/2018
- Directive 2001/83/EC. Link.
- EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines. Link.
- The European Pharmacopoeia. https://pheur.edqm.eu/home
- Council Directive 2013/59/Euratom. Link.
- Faivre-Chauvet, A., Bourdeau, C. and Bourgeois, M. Radiopharmaceutical good practices: Regulation between hospital and industry. Front. Nucl. Med., 07 September 2022. Sec. Radiopharmacy and Radiochemistry Volume 2 – 2022 | https://doi.org/10.3389/fnume.2022.990330
- Regulation (EU) No 1235/2010. Link.
- Samineni, D., Venkatakrishnan, K., Othman, A., et al. Dose Optimization in Oncology Drug Development: An International Consortium for Innovation and Quality in Pharmaceutical Development White Paper. Clinical Pharmacology & Therapeutics: Volume 116, Issue 3. Getting the Dosage Right: 479-874. September 2024. https://doi.org/10.1002/cpt.3298
- Poli G.L., Colinet, N. NEW CRP: Dosimetry in Radiopharmaceutical Therapy for Personalized Patient Treatment (E23005). IAEA International Atomic Energy Agency. Link.
- What is Nuclear Medicine? Link.
AUTHORED BY

Principal SME, Pharmaceutical Services
Maurice Bancsi
Other Related Articles

26 Jun, 2026

05 Jun, 2026


07 May, 2026

10 Apr, 2026

