

EMA Roadmap Update: PMS Capabilities and the End of the Legacy Era
The UK Clinical Trials Regulation Overhaul

23 Jun, 2025
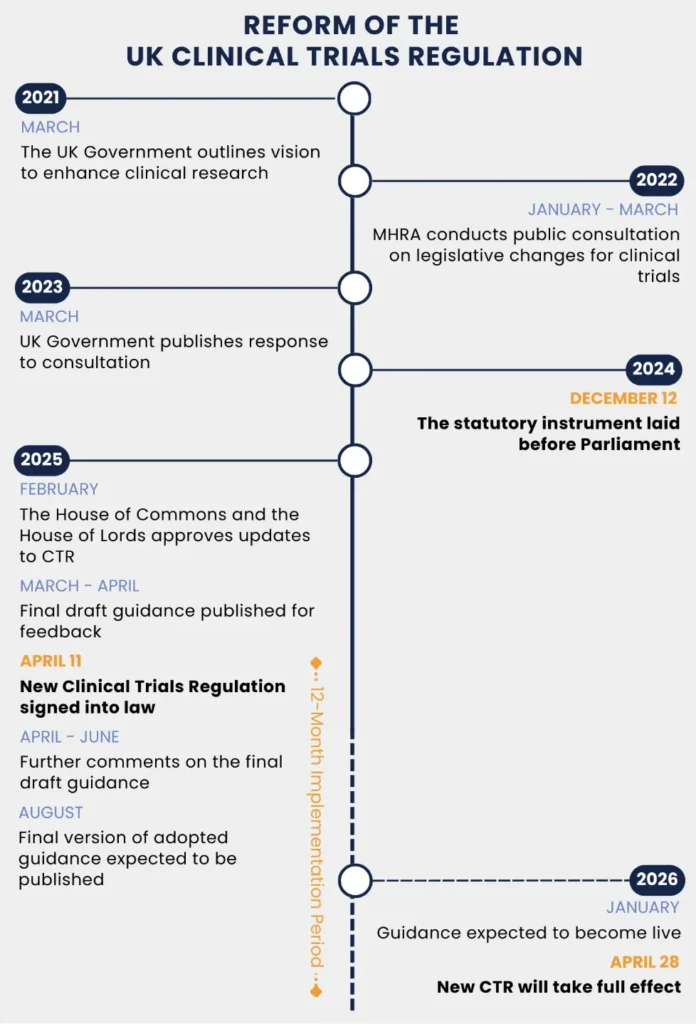
The United Kingdom is undergoing its most significant reform of clinical trials regulations (CTR) in over two decades. With the ‘Medicines for Human Use (Clinical Trials) (Amendment) Regulations 2024’, the UK is introducing a more agile, transparent, and patient-focused approach to clinical research. Between 2022 and 2023, the MHRA – in collaboration with the Department of Health in Northern Ireland – undertook a consultation period around a set of proposals to improve and strengthen the UK clinical trials legislation. This resulted in new legislation laid before Parliament on December 12, 2024, and on April 11, 2025, these new regulations have been signed into law. As there will be a 12-month implementation period, this means that the new regulations would take full effect on April 10, 2026. However, due to a technical issue during the final processing of the Clinical Trial Statutory Instrument (SI) within the digital legislation platform and the need to re-sign the SI, this date has changed to 28 April 2026.This blog outlines the main changes that are introduced by the new CTR in the UK.
Figure 1 Timeline of the UK Clinical Trial Regulation Reform
Why this change in regulation matters?
The revision of the UK’s CTR marks a pivotal shift to revitalizing the country’s research landscape. The previous regulatory framework largely aligned with the EU Clinical Trials Directive (2001/20/EC) and was often seen as inflexible and administratively burdensome. By revising this legislation, a modernized, streamlined and proportionate regulatory environment will be created that encourages innovation while protecting participants.
Leveraging the UK’s autonomy post-Brexit and lessons learned from the COVID-19 pandemic, the renewal of the clinical trials legislative framework aims to make the UK a more attractive and flexible place for clinical research. For sponsors, CROs and investigators, this reform offers a significant opportunity for faster start-up times, greater regulatory clarity and an environment that actively supports new trial designs.
Key Features of the New Regulation
The reform’s key features are to ensure that patients and their safety are at the focus of all clinical trials and bring the benefits of clinical trials to everyone. Furthermore, duplication and unnecessary delays will be cut, while maintaining robust oversight of the safety of trials and creating a proportionate and flexible regulatory environment and thereby favoring the UK as a destination for international trials. Finally, a framework will be provided that is streamlined, agile and responsive to innovation. These aims are facilitated by changes to different aspects of the regulation, of which the most important ones are outlined below.
Approval process – Integrated Regulatory and Ethics Review
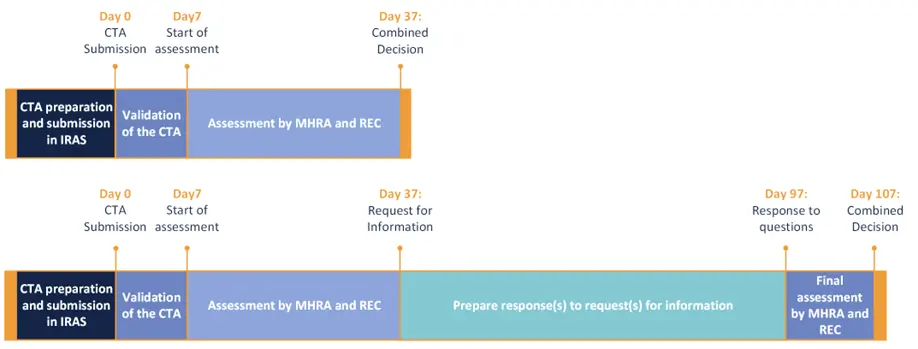
To streamline the approval process, a combined review process is introduced for clinical trial applications, bringing together the MHRA and Research Ethics Committees (RECs) into a single, coordinated assessment. The application for this combined review is to be made through the Integrated Research Application System (IRAS).
Once a clinical trial application is validated (which can take up to 7 calendar days), the combined decision will be issued within 30 calendar days. When further information is requested by MHRA/REC, the applicant has a maximum of 60 calendar days to respond. Within 10 calendar days following receipt of the response, a decision will be available. These new timelines can significantly reduce approval timelines in the UK.
Figure 2 Maximum timelines for combined CTA assessment (with and without request for information) by MHRA and REC under the new Clinical Trials Regulation
Although the combined review process will become the norm, there will be an option for separate submissions to MHRA and REC. However, this is generally discouraged and only accepted in exceptional cases. For example, when there is a serious safety issue in an ongoing clinical trial, an urgent substantial modification may need to be submitted to MHRA first to avoid delays, followed by a separate submission to the REC. Other examples include when early scientific advice is sought from MHRA before finalizing the protocol, when MHRA authorization is required, but REC approval is not (e.g. non-interventional trials or certain Phase 0 trials), or when a non-standard development pathway is followed, like the innovative licensing and access pathway (ILAP).
Risk-Based Regulation and the Notification Scheme
At the core of the proposed changes is a commitment to proportionality. Rather than applying a one-size-fits-all model, the new framework introduces regulatory requirements tailored to the risk profile of a given trial. Amongst the most important changes is the introduction of so called notifiable clinical trials. These are studies classified as lower risk, such as those involving already approved medicines used within their licensed indications. For these type of trials, full MHRA approval will no longer be required. Instead, a simple notification will suffice, although approval from a Research Ethics Committee (REC) will still be mandatory.
In October 2023, an initial notification scheme has been introduced for eligible, low-risk Phase 3 and Phase 4 studies. Such applications are processed within 14 days, instead of the standard 30 days, aiming to reduce start-up time, without undermining patient safety. With the new CTR, MRHA aims to embed this notification scheme into legislation, promoting a more risk-proportionate and streamlined approach to clinical trial approvals. It is expected by MHRA that about 20% of UK initial clinical trial applications will be eligible for this notification scheme. An important note here is that the notification scheme only applies to initial applications and cannot be used for clinical trial modifications.
Enhanced Transparency Requirements
The new CTR embeds transparency as a regulatory requirement, aiming to increase public trust and research integrity. The transparency requirements involve a mandatory registration of all trials on a WHO-compliant public registry, like ISRCTN (International Standard Randomized Controlled Trial Number; The UK’s Clinical Study Registry, including all types of clinical studies), ClinicalTrial.gov or CTIS (Clinical Trials Information System; The EU clinical trials registry). This shall be done by the date on which the first participant is recruited, or 90 days after the clinical trial has been authorized (whichever date is earlier). In addition, a summary of the study results needs to be published on the same public registry within 12 months of study completion. It is also required to offer to share a summary of the results in a manner that is understandable to laypersons with participants (or relevant persons) in a suitable format.
These requirements will apply to all UK trials and bring clarity to sponsors on expectations for dissemination. There will be options available for sponsors to request a deferral or waiver to protect commercially confidential information, or in other exceptional circumstances, on a case-by-case basis.
Good Clinical Practice, Safety reporting and Pharmacovigilance
The proposed reforms bring the UK’s GCP requirements in line with updated ICH guidelines. This includes a focus on quality-by-design principles and risk-proportional monitoring, as outlined in ICH E6(R3). This alignment will ensure that UK clinical trial data is accepted globally, as UK clinical trials will meet international standards and interoperability.
Current UK safety reporting requirements have been criticized for generating administrative burden without enhancing participant safety. The new guidance proposes a more pragmatic approach, eliminating duplication and refocusing on meaningful, risk-driven safety surveillance. This is done by removing duplicative safety reporting requirements that do not further contribute to patient safety and includes removal of the requirement to include listings of serious adverse events and serious adverse reactions in annual safety reports. Instead, an appropriate narrative of how safety concerns have been evaluated and interpreted will be included.
Key Manufacturing Updates
The UK’s new clinical trials law introduces several significant changes to the manufacturing and assembly of trial medications. By giving non-investigational medicinal products (NIMPs) a clearer definition, it increases trust in their safety and quality. The regulation also exempts radiopharmaceuticals from the requirement for a separate Manufacturing Authorization for investigational pharmaceuticals. Finally, it allows for more flexible labeling requirements, when necessary, which promotes a more adequate approach.
Guidance
Both MHRA and HRA developed new guidance to support the conduct of clinical trials in the UK. Where the MHRA guidance is focusing on the regulatory and safety oversight aspects, the HRA guidance is covering aspects of ethics and governance in research. Although separate guidance is developed by MHRA and HRA, both parties have worked closely together to ensure consistency and a similar time scheme to enable that both guidance complement each other.
The MHRA guidance is structured around seven main areas:
1. Requesting approval for a clinical trial
2. Modifications to clinical trial approval
3. Notifiable trials and the notification scheme
4. Safety
5. Research transparency
6. Simplified means of seeking and recording consent
7. Ending a clinical trial
In due course, MHRA will provide additional guidance on other topics as labelling requirements, NIMPs, and Good Clinical Practice.
The HRA guidance covers some aspects that the MHRA guidance covers as well, but mainly addresses topics outside the scope of MHRA. The focus of the HRA guidance is on the new aspects of the legislation, clearly outlining any changes. This includes clarification on research transparency, such as how deferral requests will be handled and expectations for providing lay summaries of results to participants and their legal representatives. For low-risk trials involving only authorized medicines used within routine care, simplified consent procedures will apply, for which guidance is provided. New inclusion and diversity guidance will help ensure trials involve participants who could be affected by the research and address underrepresentation, forming the basis for an inclusion and diversity plan. Additionally, the HRA is preparing tailored guidance on public involvement, with a particular focus on addressing current gaps in Phase 1 healthy volunteer trials.
Conclusion
With the revision of the CTR, the UK is establishing itself as an international research leader. The new regime prioritizes patient/participant safety, avoids unnecessary duplication and delays in trials, and introduces a more balanced and responsive regulatory approach, especially for lower-risk trials. By reducing bureaucracy and promoting innovation, the UK aims to create a responsive and agile environment that can attract foreign clinical trials and deliver benefits to patients in a faster manner.
Further reading
AUTHORED BY
Subject Matter Expert, Regulatory Affairs
Diede van Bladel
Other Related Articles

26 Mar, 2026

10 Mar, 2026

24 Feb, 2026



06 Feb, 2026
