

Heavy Metals in Pharmaceuticals: A Critical Quality and Safety Imperative
Regulatory Considerations for Radiotherapeutics and Radiodiagnostics in the US

11 Sep, 2025
This is the 2nd blog in the series “Understanding Radiotherapeutics and Radiodiagnostics.” Read the 1st blog: Regulatory Considerations for Radiotherapeutics & Radiodiagnostics
FDA Guidance for Radiotherapeutics and Radiodiagnostics
As a single country, the US presents a relatively simple regulatory framework for RPs, compared to Europe. The Food and Drug Administration (FDA) regulates radioisotopes used in nuclear medicine procedures and studies, with various approval pathways for different types of RP products.
Late Radiation Toxicity Studies for Therapeutic RPs
A 2011 FDA guidance document (1) offers recommendations to help the industry design nonclinical studies on late radiation toxicity for therapeutic radiopharmaceuticals. The goal of these studies is to reduce the risk of late-onset radiation toxicities in clinical trials. Since other guidelines cover standard nonclinical safety studies, this one focuses specifically on late radiation safety issues unique to therapeutic radiopharmaceuticals. These concerns arise because these treatments can expose normal organs to high doses of ionizing radiation, potentially causing irreversible damage.
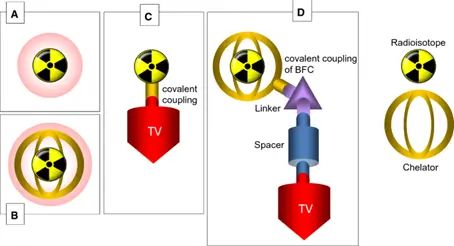
Figure 1: Examples of Radiotheranostics. A: direct injection of radionuclide, B: injection of complexed radiometal, C: covalent coupling of a radionuclide to a targeting vector (red), D: radiometal complexed by bifunctional chelator (BFC) which is covalently bound by a linker to the targeting vector and a possible spacer (3)
This guidance is not intended to address late radiation toxicity of radiobiologicals (e.g., radiolabeled monoclonal antibodies). This guidance is also not intended to apply to diagnostic radiopharmaceuticals whose low doses are not expected to elicit late radiation toxic effects.
Microdose Radiopharmaceutical Diagnostic Drugs
Another FDA guidance, focusing on microdose RP diagnostic drugs, intends to refine nonclinical study recommendations for radiodiagnostics based on their unique characteristics, which include microdosing and single or infrequent clinical use; this 2018 guidance reduces or eliminates additional toxicology requirements, and clarifies other nonclinical requirements for microdose diagnostic RPs for Phase 1–3 studies(2).
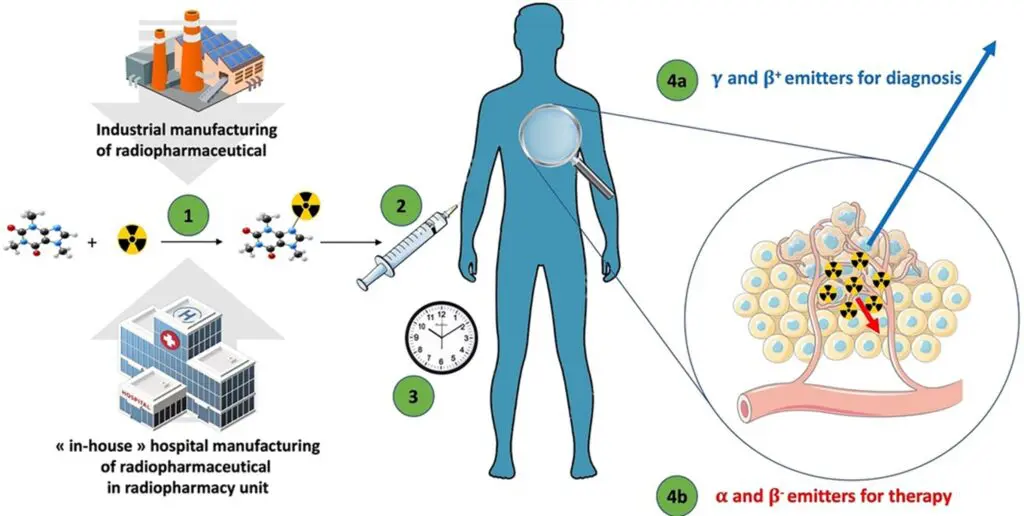
Figure 2: The general flow of microdose radiopharmaceuticals. (1) The radiopharmaceutical compound is manufactured by the industry or “in-house” hospital radiopharmacy unit under GMP or PIC/S regulation, respectively. (2) The radiopharmaceutical compound is injected into the patient in the nuclear medicine department. (3) After the elapsed time needed for the specific pharmacological distribution of the radiopharmaceutical, the radioactivity is used depending on the purpose: (4a) an emission of radioactivity outside the body for external detection (diagnostic) with γ or β+ emitters; or (4b) local irradiation for therapeutic purpose with α, β−, or Auger emitters (4).
Nuclear Regulatory Commission (NRC) Oversight
In addition to the FDA, the United States Nuclear Regulatory Commission (USNRC), established in 1975, maintains regulatory oversight of nuclear materials in the US, and devices used to administer RPs are regulated by the individual states. Ultimately, the onus of responsibility rests with the medical institution (whether a hospital or university), which must ensure that the machinery and devices meet regulatory compliance and have current licensure. The manufacturer/vendor, in turn, must have approval to produce isotopes, and must adhere to standard operating procedures (SOPs). That leaves the investigator as a “middleman” who must work with an institution that has an approved device, and with a manufacturer licensed to produce the isotopes.
A specific license is required to receive, possess, use, transfer, or acquire radioactive materials in the US. The NRC issues such licenses for possession and use of RP byproduct, source, and special nuclear material. Applications for possession and use must be submitted to one of the NRC’s regional offices. The licensed facility must follow the Division of Radioactive Material Licensing and Compliance regulations.
Radioisotope Review Committee Approval Process
One of the most important regulatory requirements in the US is that the sponsor must obtain Radioisotope Review Committee approval before submitting an RP clinical trial application to an Institutional Review Board (IRB). The US regulatory process also involves close scrutiny of the number of computed tomography (CT) scans (such as in an oncology trial), and of the number of diagnostics needed in a trial. Sponsors must also secure approval from IRBs as well as from other hospital organizations that regulate the use of radiopharmaceuticals and radiodiagnostics.
The FDA’s Radioactive Drug Research Committee (RDRC) Program
The Radioactive Drug Research Committee (RDRC) program was established by the FDA in 1975 to regulate the use of radioactive drugs in research. Basic research on radioactive drugs in humans is allowed without requiring an Investigational New Drug (IND) application, as long as specific conditions are met.
The research must focus on obtaining fundamental scientific knowledge about drug metabolism, human physiology, or biochemistry, without aiming for therapeutic or diagnostic purposes or assessing drug safety and effectiveness. Studies must be approved by an FDA-approved RDRC and meet requirements such as qualified investigators, licensed facilities, informed consent, quality assurance, IRB approval, and adverse event reporting.
Additionally, the administered radioactive drug must have no detectable pharmacologic effect, and the radiation dose must be justified by the study’s scientific value while staying within regulatory limits.
How Celegence Supports Radiopharmaceutical Sponsors
Celegence’s team of subject matter experts in regulatory strategy for pharmaceuticals and medical devices ensures that both country-specific and market-wide regulatory requirements are met. We provide sponsors with tailored guidance and develop strategic roadmaps to maximize the efficiency of radiodiagnostics and radiotherapeutics in clinical trials. Most importantly, our experts act as on-the-ground partners, helping sponsors navigate the complexities of the regulatory landscape with confidence.
Contact us today at info@celegence.com to learn how our experts can help you navigate FDA, NRC, and RDRC requirements for radiopharmaceutical development with confidence and compliance.
References
- Guidance for Industry: Nonclinical Evaluation of Late Radiation Toxicity of Therapeutic Radiopharmaceuticals. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER); 2011. https://www.fda.gov/media/72237/download
- Microdose Radiopharmaceutical Diagnostic Drugs: Nonclinical Study Recommendations – Guidance for Industry. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER); 2018. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/microdose-radiopharmaceutical-diagnostic-drugs-nonclinical-study-recommendations
- Roesch, F., Martin, M. Radiometal-theranostics: the first 20 years*. J Radioanal Nucl Chem 332, 1557–1576 (2023). https://doi.org/10.1007/s10967-022-08624-3
- Faivre-Chauvet, A., Bourdeau, C. and Bourgeois, M. Radiopharmaceutical good practices: Regulation between hospital and industry. Front. Nucl. Med., 07 September 2022. Sec. Radiopharmacy and Radiochemistry Volume 2 – 2022 | https://doi.org/10.3389/fnume.2022.990330
AUTHORED BY

Principal SME, Pharmaceutical Services
Maurice Bancsi
Other Related Articles


26 Mar, 2026

10 Mar, 2026

24 Feb, 2026


