
PMCF Under EU MDR: Lessons from HCP Surveys and Real-World Data Collection
Implementing and Maintaining PMS & Performance Evaluation under the IVDR – Webinar Q&A

24 Nov, 2022
[fusion_builder_container hundred_percent=”no” hundred_percent_height=”no” hundred_percent_height_scroll=”no” hundred_percent_height_center_content=”yes” equal_height_columns=”no” menu_anchor=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” status=”published” publish_date=”” class=”” id=”” border_color=”” border_style=”solid” margin_top=”0″ margin_bottom=”” padding_top=”0px” padding_right=”” padding_bottom=”” padding_left=”” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”” background_image=”” background_position=”center center” background_repeat=”no-repeat” fade=”no” background_parallax=”none” enable_mobile=”no” parallax_speed=”0.3″ background_blend_mode=”none” video_mp4=”” video_webm=”” video_ogv=”” video_url=”” video_aspect_ratio=”16:9″ video_loop=”yes” video_mute=”yes” video_preview_image=”” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ type=”legacy”][fusion_builder_row][fusion_builder_column type=”1_1″ layout=”1_1″ spacing=”yes” center_content=”no” link=”” target=”_self” min_height=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” class=”” id=”” background_image_id=”” hover_type=”none” border_color=”” border_style=”solid” border_position=”all” border_radius_top_left=”” border_radius_top_right=”” border_radius_bottom_right=”” border_radius_bottom_left=”” box_shadow=”no” box_shadow_vertical=”” box_shadow_horizontal=”” box_shadow_blur=”0″ box_shadow_spread=”0″ box_shadow_color=”” box_shadow_style=”” padding_top=”0″ padding_right=”” padding_bottom=”” padding_left=”” margin_top=”0px” margin_bottom=”0px” background_type=”single” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”” background_image=”” background_position=”left top” background_repeat=”no-repeat” background_blend_mode=”none” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=”” filter_type=”regular” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ last=”true” border_sizes_top=”0″ border_sizes_bottom=”0″ border_sizes_left=”0″ border_sizes_right=”0″ first=”true”][fusion_imageframe image_id=”15428|full” aspect_ratio=”” custom_aspect_ratio=”100″ aspect_ratio_position=”” skip_lazy_load=”” lightbox=”no” gallery_id=”” lightbox_image=”” lightbox_image_id=”” alt=”Implementing Maintaining PMS Performance Evaluation IVDR – Q&A – Celegence” link=”” linktarget=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” max_width=”” sticky_max_width=”” align_medium=”none” align_small=”none” align=”none” mask=”” custom_mask=”” mask_size=”” mask_custom_size=”” mask_position=”” mask_custom_position=”” mask_repeat=”” style_type=”” blur=”” stylecolor=”” hue=”” saturation=”” lightness=”” alpha=”” hover_type=”none” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” bordersize=”” bordercolor=”” borderradius=”” z_index=”” caption_style=”off” caption_align_medium=”none” caption_align_small=”none” caption_align=”none” caption_title=”” caption_text=”” caption_title_tag=”2″ fusion_font_family_caption_title_font=”” fusion_font_variant_caption_title_font=”” caption_title_size=”” caption_title_line_height=”” caption_title_letter_spacing=”” caption_title_transform=”” caption_title_color=”” caption_background_color=”” fusion_font_family_caption_text_font=”” fusion_font_variant_caption_text_font=”” caption_text_size=”” caption_text_line_height=”” caption_text_letter_spacing=”” caption_text_transform=”” caption_text_color=”” caption_border_color=”” caption_overlay_color=”” caption_margin_top=”” caption_margin_right=”” caption_margin_bottom=”” caption_margin_left=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=”” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″]https://www.celegence.com/wp-content/uploads/2022/11/Implementing-Maintaining-PMS-Performance-Evaluation-IVDR-QA-Celegence.jpg[/fusion_imageframe][fusion_separator style_type=”none” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” class=”” id=”” sep_color=”” top_margin=”30px” bottom_margin=”” border_size=”” icon=”” icon_circle=”” icon_circle_color=”” width=”” alignment=”center” /][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”1″ animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” hue=”” saturation=”” lightness=”” alpha=”” animated_text_color=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]Implementing and Maintaining PMS & Performance Evaluation under the IVDR – Webinar Q&A[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]
Celegence, a Global Regulatory Affairs Services & Technology company for the Life Sciences Industry, partnered with Q1 Productions in September 2022 to offer a webcast titled “Implementing and Maintaining PMS and Performance Evaluation under the IVDR”.
By replaying this webinar, you will have an opportunity to understand how the PMS process interoperates with other systems including Performance Evaluation, Risk Management, and Post Market Performance Follow-Up to continuously update critical elements of the Technical Documentation.
If you were not able to attend this webinar, here’s a link where you can watch the on-demand version embedded below. We are here to address any other questions that you may have related to the webinar or questions.
This post contains the Q&A session from the webinar – giving you the answers to the most common questions about implementing and maintaining PMS and Performance Evaluation under the IVDR.
[/fusion_text][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]
A full transcript of James Shearn’s presentation is available to download (and to read below) and just press play to watch the clip now.
[/fusion_text][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”default” rule_size=”” rule_color=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” font_size=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” line_height=”” letter_spacing=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””][/fusion_text][/fusion_builder_column][/fusion_builder_row][/fusion_builder_container][fusion_global id=”5197″][fusion_builder_container hundred_percent=”no” hundred_percent_height=”no” hundred_percent_height_scroll=”no” hundred_percent_height_center_content=”yes” equal_height_columns=”no” menu_anchor=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” status=”published” publish_date=”” class=”” id=”” border_color=”” border_style=”solid” margin_top=”” margin_bottom=”” padding_top=”” padding_right=”” padding_bottom=”” padding_left=”” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”” background_image=”” background_position=”center center” background_repeat=”no-repeat” fade=”no” background_parallax=”none” enable_mobile=”no” parallax_speed=”0.3″ background_blend_mode=”none” video_mp4=”” video_webm=”” video_ogv=”” video_url=”” video_aspect_ratio=”16:9″ video_loop=”yes” video_mute=”yes” video_preview_image=”” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ admin_toggled=”no” type=”legacy”][fusion_builder_row][fusion_builder_column type=”1_1″ layout=”2_3″ spacing=”” center_content=”no” link=”” target=”_self” min_height=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” class=”” id=”” background_image_id=”” hover_type=”none” border_color=”” border_style=”solid” border_position=”all” border_radius_top_left=”” border_radius_top_right=”” border_radius_bottom_right=”” border_radius_bottom_left=”” box_shadow=”no” box_shadow_vertical=”” box_shadow_horizontal=”” box_shadow_blur=”0″ box_shadow_spread=”0″ box_shadow_color=”” box_shadow_style=”” padding_top=”” padding_right=”” padding_bottom=”” padding_left=”” margin_top=”” margin_bottom=”” background_type=”single” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”” background_image=”” background_position=”left top” background_repeat=”no-repeat” background_blend_mode=”none” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=”” filter_type=”regular” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ last=”true” border_sizes_top=”0″ border_sizes_bottom=”0″ border_sizes_left=”0″ border_sizes_right=”0″ first=”true”][fusion_separator style_type=”none” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” class=”” id=”” sep_color=”” top_margin=”15px” bottom_margin=”” border_size=”” icon=”” icon_circle=”” icon_circle_color=”” width=”” alignment=”center” /][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”2″ animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” hue=”” saturation=”” lightness=”” alpha=”” animated_text_color=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]Q: If any clinical evidence that is an “EQA” data clinical sample. For example, IQM-positive for herpes simplex virus is available. How can I produce clinical evidence of the validity of my device?[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]A: It falls into the one element of what you would pull into your clinical performance report. You would bring that in as a reference, and you would discuss that as an element of data in the context of any other supporting information you have. For example, something that you identified in a literature review, can be pulled in and reviewed within the clinical performance report and then within a performance evaluation as a result.[/fusion_text][fusion_imageframe image_id=”15429|full” aspect_ratio=”” custom_aspect_ratio=”100″ aspect_ratio_position=”” skip_lazy_load=”” lightbox=”no” gallery_id=”” lightbox_image=”” lightbox_image_id=”” alt=”CAPTIS Documentation Notified Body Format – Celegence” link=”” linktarget=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” max_width=”” sticky_max_width=”” align_medium=”none” align_small=”none” align=”none” mask=”” custom_mask=”” mask_size=”” mask_custom_size=”” mask_position=”” mask_custom_position=”” mask_repeat=”” style_type=”” blur=”” stylecolor=”” hue=”” saturation=”” lightness=”” alpha=”” hover_type=”none” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” bordersize=”” bordercolor=”” borderradius=”” z_index=”” caption_style=”off” caption_align_medium=”none” caption_align_small=”none” caption_align=”none” caption_title=”” caption_text=”” caption_title_tag=”2″ fusion_font_family_caption_title_font=”” fusion_font_variant_caption_title_font=”” caption_title_size=”” caption_title_line_height=”” caption_title_letter_spacing=”” caption_title_transform=”” caption_title_color=”” caption_background_color=”” fusion_font_family_caption_text_font=”” fusion_font_variant_caption_text_font=”” caption_text_size=”” caption_text_line_height=”” caption_text_letter_spacing=”” caption_text_transform=”” caption_text_color=”” caption_border_color=”” caption_overlay_color=”” caption_margin_top=”” caption_margin_right=”” caption_margin_bottom=”” caption_margin_left=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=”” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″]https://www.celegence.com/wp-content/uploads/2022/11/CAPTIS-Documentation-Notified-Body-Format-Celegence.jpg[/fusion_imageframe][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”3″ animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” hue=”” saturation=”” lightness=”” alpha=”” animated_text_color=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]Q: Must the intended use be listed on the various reports, or can the report reference the DHF which contains the intended use?[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]
A: It is not a requirement for you to have intended use in your document, but I highly recommend you make one. It means you don’t have to write it out all the time. You can then reference that from your other sections of the documentation.
[/fusion_text][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”3″ animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” hue=”” saturation=”” lightness=”” alpha=”” animated_text_color=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]Q: How do you demonstrate State of the Art (SoTA). Would you recommend preparing a separate document for SoTA as it is also repeated content or are there other solutions?[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]A: You could do it either way. You are required to describe the state of the art within the performance evaluation process. However, you could do that as a separate document to reference it, or you could do it in the same document. I think it would be very much dependent upon you and your business and how that works best in your QMS.[/fusion_text][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”3″ animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” hue=”” saturation=”” lightness=”” alpha=”” animated_text_color=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]Q: Do you think if we defined PMPF as not applicable, if our product has already been sold for many years and has international standards for the product?[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]A: It depends. If you have a lot of data in the market about a product, and you do not see a massive amount of change through your PMS, then it’s going to be easier for you to make the argument that it’s not required. It’s one of those things that you have to come back to every year or every time when running your cycle. If you are not finding anything new in your cycle, then you may be able to continue to make that argument. You have to be aware, that your post-market surveillance cycle may throw up things that you were unaware of before, and that may change what’s required. You may, as the output of a cycle, find that you have a new piece of information that requires you to go and investigate a particular aspect and potentially do some active work like running a study. You have to answer that question every time you’re in the cycle.[/fusion_text][/fusion_builder_column][/fusion_builder_row][/fusion_builder_container][fusion_builder_container hundred_percent=”no” hundred_percent_height=”no” hundred_percent_height_scroll=”no” hundred_percent_height_center_content=”yes” equal_height_columns=”no” menu_anchor=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” status=”published” publish_date=”” class=”” id=”” border_color=”” border_style=”solid” margin_top=”” margin_bottom=”” padding_top=”” padding_right=”” padding_bottom=”” padding_left=”” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”” background_image=”” background_position=”center center” background_repeat=”no-repeat” fade=”no” background_parallax=”none” enable_mobile=”no” parallax_speed=”0.3″ background_blend_mode=”none” video_mp4=”” video_webm=”” video_ogv=”” video_url=”” video_aspect_ratio=”16:9″ video_loop=”yes” video_mute=”yes” video_preview_image=”” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ admin_toggled=”no” type=”legacy”][fusion_builder_row][fusion_builder_column type=”1_1″ layout=”2_3″ spacing=”” center_content=”no” link=”” target=”_self” min_height=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” class=”” id=”” background_image_id=”” hover_type=”none” border_color=”” border_style=”solid” border_position=”all” border_radius_top_left=”” border_radius_top_right=”” border_radius_bottom_right=”” border_radius_bottom_left=”” box_shadow=”no” box_shadow_vertical=”” box_shadow_horizontal=”” box_shadow_blur=”0″ box_shadow_spread=”0″ box_shadow_color=”” box_shadow_style=”” padding_top=”” padding_right=”” padding_bottom=”” padding_left=”” margin_top=”” margin_bottom=”” background_type=”single” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”” background_image=”” background_position=”left top” background_repeat=”no-repeat” background_blend_mode=”none” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=”” filter_type=”regular” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ last=”true” border_sizes_top=”0″ border_sizes_bottom=”0″ border_sizes_left=”0″ border_sizes_right=”0″ first=”true”][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”3″ animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” hue=”” saturation=”” lightness=”” alpha=”” animated_text_color=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]Q: How many details are required for complaints? Is it just a general number or do we need to talk about each failure mode?[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]
A: It will largely depend on the kind of business you are in and how many complaints you get. If you’re in a business that gets a lot of feedback, you will have much more numeric summaries. If it’s only like ones and twos, you may find that kind of thing. If you’re getting 3, 4, 5, or 10 complaints or reports in a given cycle, then you may end up providing more detail. It depends on the PMS reports, I would tend to go more towards summarization than extreme detail. Then you might be looking at what your top issues are when you go into that cross-functional review. Then that’s going to inform the areas where you might potentially want to go and do further investigation to get a better understanding of those particular aspects or potentially update your design to address those issues.
[/fusion_text][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”3″ animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” hue=”” saturation=”” lightness=”” alpha=”” animated_text_color=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]
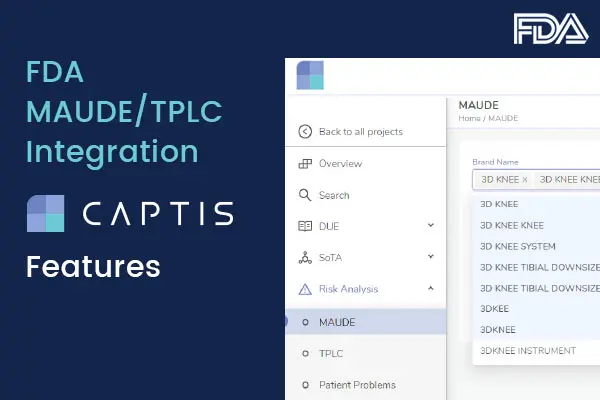
Q: How does CAPTIS™ generate the documentation to be delivered to the notified body and in which format?
[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]
A: CAPTIS™ is a tool that allows you to tag and track different sections of text as you compile that documentation that then needs to be transferred into whatever sort of document control process you are using yourselves. It’s probably very dependent upon you and your business.
[/fusion_text][/fusion_builder_column][/fusion_builder_row][/fusion_builder_container][fusion_global id=”5197″][fusion_builder_container hundred_percent=”no” hundred_percent_height=”no” hundred_percent_height_scroll=”no” hundred_percent_height_center_content=”yes” equal_height_columns=”no” menu_anchor=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” status=”published” publish_date=”” class=”” id=”” border_color=”” border_style=”solid” margin_top=”” margin_bottom=”” padding_top=”” padding_right=”” padding_bottom=”” padding_left=”” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”” background_image=”” background_position=”center center” background_repeat=”no-repeat” fade=”no” background_parallax=”none” enable_mobile=”no” parallax_speed=”0.3″ background_blend_mode=”none” video_mp4=”” video_webm=”” video_ogv=”” video_url=”” video_aspect_ratio=”16:9″ video_loop=”yes” video_mute=”yes” video_preview_image=”” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ admin_toggled=”no” type=”legacy”][fusion_builder_row][fusion_builder_column type=”1_1″ layout=”2_3″ spacing=”” center_content=”no” link=”” target=”_self” min_height=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” class=”” id=”” background_image_id=”” hover_type=”none” border_color=”” border_style=”solid” border_position=”all” border_radius_top_left=”” border_radius_top_right=”” border_radius_bottom_right=”” border_radius_bottom_left=”” box_shadow=”no” box_shadow_vertical=”” box_shadow_horizontal=”” box_shadow_blur=”0″ box_shadow_spread=”0″ box_shadow_color=”” box_shadow_style=”” padding_top=”” padding_right=”” padding_bottom=”” padding_left=”” margin_top=”” margin_bottom=”” background_type=”single” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”” background_image=”” background_position=”left top” background_repeat=”no-repeat” background_blend_mode=”none” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=”” filter_type=”regular” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ last=”true” border_sizes_top=”0″ border_sizes_bottom=”0″ border_sizes_left=”0″ border_sizes_right=”0″ first=”true”][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”3″ animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” hue=”” saturation=”” lightness=”” alpha=”” animated_text_color=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]Q: Do we need to conduct analytical studies for legacy products?[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]
A: It depends on how much evidence you have. You need to go through your performance evaluation plan and establish a strategy to review the data that you have. Additionally, you need to understand your gaps, which will help you decide your following action. You need to identify your gaps, and that will then tell you whether you need to perform additional studies.
[/fusion_text][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”3″ animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” hue=”” saturation=”” lightness=”” alpha=”” animated_text_color=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]Q: For controls, do we need clinical performance reports as per IVDR? (These are not essays and do not aid in diagnostics or measure the analyte, so we can’t avoid writing CPRs for controls, which are third party independent controls)[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]
A: You can justify that. You can make it just because your control doesn’t have a clinical application in itself.
[/fusion_text][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”3″ animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” hue=”” saturation=”” lightness=”” alpha=”” animated_text_color=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]Q: There is a great degree of overlap in ANNEX XIII of IVDR, between PMPF and PER. Any strategy to manage the overlap?[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]
A: When conducting your literature reviews, do you need to do some of the performance evaluation and some of the PMS? If we take that as an example, you can help yourself by synchronizing those and doing one set of literature reviews that meet the requirements of both.
[/fusion_text][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”3″ animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” hue=”” saturation=”” lightness=”” alpha=”” animated_text_color=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]Q: Can we write SoTA in SVR documents as some of the companies are asking for the same?[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]A: For the requirements, it has to be in the right place. If you were to include it in SVR as well and then references, that would be alright. But you need to make sure that each of your deliverables, each of the 3 reports has all the sections in it that are required by the regulation because they need to be there for part of the review by the notified body.[/fusion_text][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”3″ animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” hue=”” saturation=”” lightness=”” alpha=”” animated_text_color=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]Q: Can we replace sales volume with another parameter such as number of tests, patients, samples managed?[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]
A: You need to select what is going to be the most appropriate sort of data depending on your kind of device. It will be reviewed by your notified body, so they will have a view as to whether that is adequate or not. When you look into things like usage frequency, which can be extremely different between different kinds of devices. When you’re thinking about data associated with complaints, you need to think about how quickly your devices get on the market from when you manufacture them. You need to look at individual case for case basis. If you devise a strategy, check that with your notified body as you get to that stage.
[/fusion_text][/fusion_builder_column][/fusion_builder_row][/fusion_builder_container][fusion_builder_container hundred_percent=”no” hundred_percent_height=”no” hundred_percent_height_scroll=”no” hundred_percent_height_center_content=”yes” equal_height_columns=”no” menu_anchor=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” status=”published” publish_date=”” class=”” id=”” border_color=”” border_style=”solid” margin_top=”” margin_bottom=”” padding_top=”” padding_right=”” padding_bottom=”” padding_left=”” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”” background_image=”” background_position=”center center” background_repeat=”no-repeat” fade=”no” background_parallax=”none” enable_mobile=”no” parallax_speed=”0.3″ background_blend_mode=”none” video_mp4=”” video_webm=”” video_ogv=”” video_url=”” video_aspect_ratio=”16:9″ video_loop=”yes” video_mute=”yes” video_preview_image=”” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ admin_toggled=”no” type=”legacy”][fusion_builder_row][fusion_builder_column type=”1_1″ layout=”2_3″ spacing=”” center_content=”no” link=”” target=”_self” min_height=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” class=”” id=”” background_image_id=”” hover_type=”none” border_color=”” border_style=”solid” border_position=”all” border_radius_top_left=”” border_radius_top_right=”” border_radius_bottom_right=”” border_radius_bottom_left=”” box_shadow=”no” box_shadow_vertical=”” box_shadow_horizontal=”” box_shadow_blur=”0″ box_shadow_spread=”0″ box_shadow_color=”” box_shadow_style=”” padding_top=”” padding_right=”” padding_bottom=”” padding_left=”” margin_top=”” margin_bottom=”” background_type=”single” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”” background_image=”” background_position=”left top” background_repeat=”no-repeat” background_blend_mode=”none” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=”” filter_type=”regular” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ last=”true” border_sizes_top=”0″ border_sizes_bottom=”0″ border_sizes_left=”0″ border_sizes_right=”0″ first=”true”][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”3″ animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” hue=”” saturation=”” lightness=”” alpha=”” animated_text_color=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]Q: If we can’t find the relative peer-to-peer literature of our brand product and we can only search the competitor’s product to the leading brand. Will it be a problem in scientific validity?[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]
A: It will depend on how similar your product is, and can you demonstrate that? If you have two of the same products at work, then you can start to make some of that argument. If they work differently, that will be harder to make. It’s about building that evidence, putting that together, and making the argument in your scientific literature report to a credible extent.
[/fusion_text][/fusion_builder_column][/fusion_builder_row][/fusion_builder_container][fusion_global id=”5197″][fusion_builder_container hundred_percent=”no” hundred_percent_height=”no” hundred_percent_height_scroll=”no” hundred_percent_height_center_content=”yes” equal_height_columns=”no” menu_anchor=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” status=”published” publish_date=”” class=”” id=”” border_color=”” border_style=”solid” margin_top=”” margin_bottom=”” padding_top=”” padding_right=”” padding_bottom=”” padding_left=”” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”” background_image=”” background_position=”center center” background_repeat=”no-repeat” fade=”no” background_parallax=”none” enable_mobile=”no” parallax_speed=”0.3″ background_blend_mode=”none” video_mp4=”” video_webm=”” video_ogv=”” video_url=”” video_aspect_ratio=”16:9″ video_loop=”yes” video_mute=”yes” video_preview_image=”” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ admin_toggled=”no” type=”legacy”][fusion_builder_row][fusion_builder_column type=”1_1″ layout=”2_3″ spacing=”” center_content=”no” link=”” target=”_self” min_height=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” class=”” id=”” background_image_id=”” hover_type=”none” border_color=”” border_style=”solid” border_position=”all” border_radius_top_left=”” border_radius_top_right=”” border_radius_bottom_right=”” border_radius_bottom_left=”” box_shadow=”no” box_shadow_vertical=”” box_shadow_horizontal=”” box_shadow_blur=”0″ box_shadow_spread=”0″ box_shadow_color=”” box_shadow_style=”” padding_top=”” padding_right=”” padding_bottom=”” padding_left=”” margin_top=”” margin_bottom=”” background_type=”single” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”” background_image=”” background_position=”left top” background_repeat=”no-repeat” background_blend_mode=”none” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=”” filter_type=”regular” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ last=”true” border_sizes_top=”0″ border_sizes_bottom=”0″ border_sizes_left=”0″ border_sizes_right=”0″ first=”true”][fusion_title title_type=”text” rotation_effect=”bounceIn” display_time=”1200″ highlight_effect=”circle” loop_animation=”off” highlight_width=”9″ highlight_top_margin=”0″ before_text=”” rotation_text=”” highlight_text=”” after_text=”” title_link=”off” link_url=”” link_target=”_self” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” content_align_medium=”” content_align_small=”” content_align=”left” size=”4″ font_size=”” animated_font_size=”” fusion_font_family_title_font=”” fusion_font_variant_title_font=”” line_height=”” letter_spacing=”” text_shadow=”no” text_shadow_vertical=”” text_shadow_horizontal=”” text_shadow_blur=”0″ text_shadow_color=”” margin_top_medium=”” margin_right_medium=”” margin_bottom_medium=”” margin_left_medium=”” margin_top_small=”” margin_right_small=”” margin_bottom_small=”” margin_left_small=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” margin_top_mobile=”” margin_bottom_mobile=”” text_color=”” animated_text_color=”” gradient_font=”no” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ highlight_color=”” style_type=”default” sep_color=”” link_color=”” link_hover_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]Full Spectrum Services for Medical Device and IVD Manufacturers[/fusion_title][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”” rule_size=”” rule_color=”” hue=”” saturation=”” lightness=”” alpha=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” font_size=”” line_height=”” letter_spacing=”” text_transform=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]
Celegence can provide you with:
- Experience and resources to help manufacturers transition their QMS
- Templates for key PMS and Performance Evaluation deliverables
- The CAPTIS™ technology solution to aid in PMS Documentation Maintenance, including Systematic Literature Review
Celegence’s comprehensive support for creating and maintaining required documentation can help ensure your organization’s IVDR compliance. Contact us at info@celegence.com, contact us online or read more about Celegence’s IVDR services.
[/fusion_text][/fusion_builder_column][/fusion_builder_row][/fusion_builder_container][fusion_builder_container hundred_percent=”no” hundred_percent_height=”no” hundred_percent_height_scroll=”no” hundred_percent_height_center_content=”yes” equal_height_columns=”no” menu_anchor=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” status=”published” publish_date=”” class=”” id=”” border_color=”” border_style=”solid” margin_top=”” margin_bottom=”” padding_top=”” padding_right=”” padding_bottom=”40px” padding_left=”” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”” background_image=”” background_position=”center center” background_repeat=”no-repeat” fade=”no” background_parallax=”none” enable_mobile=”no” parallax_speed=”0.3″ background_blend_mode=”none” video_mp4=”” video_webm=”” video_ogv=”” video_url=”” video_aspect_ratio=”16:9″ video_loop=”yes” video_mute=”yes” video_preview_image=”” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ type=”legacy”][fusion_builder_row][fusion_builder_column type=”1_1″ layout=”1_1″ spacing=”” center_content=”no” link=”” target=”_self” min_height=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” class=”” id=”” background_image_id=”” hover_type=”none” border_color=”#eff2f2″ border_style=”solid” border_position=”all” border_radius_top_left=”10px” border_radius_top_right=”10px” border_radius_bottom_right=”10px” border_radius_bottom_left=”10px” box_shadow=”yes” box_shadow_vertical=”5%” box_shadow_horizontal=”5%” box_shadow_blur=”28″ box_shadow_spread=”2″ box_shadow_color=”#f1f6f8″ box_shadow_style=”” padding_top=”” padding_right=”0px” padding_bottom=”” padding_left=”20px” margin_top=”0px” margin_bottom=”0px” background_type=”single” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_color=”#f6f8f8″ background_image=”” background_position=”left top” background_repeat=”no-repeat” background_blend_mode=”none” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=”” filter_type=”regular” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ last=”true” border_sizes_top=”0″ border_sizes_bottom=”0″ border_sizes_left=”0″ border_sizes_right=”0″ first=”true”][fusion_builder_row_inner][fusion_builder_column_inner type=”1_1″ layout=”1_1″ spacing=”2%” center_content=”yes” hover_type=”none” link=”” target=”_self” min_height=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” class=”” id=”” border_color=”#eff2f2″ border_style=”solid” border_position=”all” border_radius_top_left=”” border_radius_top_right=”” border_radius_bottom_right=”” border_radius_bottom_left=”” box_shadow=”no” box_shadow_vertical=”” box_shadow_horizontal=”” box_shadow_blur=”0″ box_shadow_spread=”0″ box_shadow_color=”” box_shadow_style=”” padding_top=”10px” padding_right=”20px” padding_bottom=”0px” padding_left=”20px” margin_top=”0px” margin_bottom=”0px” background_type=”single” background_color=”” gradient_start_color=”” gradient_end_color=”” gradient_start_position=”0″ gradient_end_position=”100″ gradient_type=”linear” radial_direction=”center center” linear_angle=”180″ background_image=”” background_position=”left top” background_repeat=”no-repeat” background_blend_mode=”none” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=”” filter_type=”regular” filter_hue=”0″ filter_saturation=”100″ filter_brightness=”100″ filter_contrast=”100″ filter_invert=”0″ filter_sepia=”0″ filter_opacity=”100″ filter_blur=”0″ filter_hue_hover=”0″ filter_saturation_hover=”100″ filter_brightness_hover=”100″ filter_contrast_hover=”100″ filter_invert_hover=”0″ filter_sepia_hover=”0″ filter_opacity_hover=”100″ filter_blur_hover=”0″ last=”true” align_content=”center” border_sizes_top=”0″ border_sizes_bottom=”0″ border_sizes_left=”0″ border_sizes_right=”0″ first=”true”][fusion_text columns=”” column_min_width=”” column_spacing=”” rule_style=”default” rule_size=”” rule_color=”” content_alignment_medium=”” content_alignment_small=”” content_alignment=”” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” sticky_display=”normal,sticky” class=”” id=”” margin_top=”” margin_right=”” margin_bottom=”” margin_left=”” font_size=”” fusion_font_family_text_font=”” fusion_font_variant_text_font=”” line_height=”” letter_spacing=”” text_color=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””]
[fusion_fontawesome icon=”fa-info-circle fas” size=”35px” flip=”” rotate=”” spin=”no” alignment=”left” hide_on_mobile=”small-visibility,medium-visibility,large-visibility” class=”” id=”” margin_top=”” margin_right=”10px” margin_bottom=”” margin_left=”” circle=”no” iconcolor=”#2da9e0″ circlecolor=”” circlebordercolor=”” animation_type=”” animation_direction=”left” animation_speed=”0.3″ animation_offset=””][/fusion_fontawesome]
For more information on how we can help you with EU MDR requirements reach out to us at info@celegence.com, contact us online or read more about Celegence’s medical device services.
[/fusion_text][/fusion_builder_column_inner][/fusion_builder_row_inner][/fusion_builder_column][/fusion_builder_row][/fusion_builder_container]
Other Related Articles

29 Jul, 2026

22 Jul, 2026

21 Jul, 2026

08 Jul, 2026

17 Jun, 2026

13 May, 2026
